From Clinical Notes to Clinical Truth: AI-Powered HPO Phenotype Mapping for Precision Variant Prioritization
Authors: Kartik Pal, Dr. Varun Venkat, Dr. Rahila Sardar · Published: 2026 · Reading time: ~12 min
Abstract
Phenotype-driven variant prioritization is a cornerstone of clinical genomic diagnosis, yet its accuracy is fundamentally constrained by the quality of Human Phenotype Ontology (HPO) term mapping. In routine clinical practice, phenotypic data are captured as unstructured natural language, introducing substantial heterogeneity that limits reliable HPO normalization. This whitepaper describes the HPO Phenotype Matcher integrated within the Vgen23 clinical genomics platform, an AI-powered pipeline combining large language model (LLM)-based clinical term extraction with vector-semantic similarity search against the HPO knowledge base. The system employs a clinician-in-the-loop validation layer to ensure phenotypic accuracy before downstream integration with variant annotation, ACMG/AMP pathogenicity scoring, and gene–disease association databases. By transforming free-text clinical descriptions into structured, ontology-grounded phenotype profiles, the Vgen23 platform enables high-precision, explainable variant prioritization that is directly aligned with the patient’s clinical presentation.
1. Introduction
The rapid proliferation of next-generation sequencing (NGS) in clinical practice has transformed the diagnostic landscape for rare and complex genetic disorders. Whole-exome sequencing (WES) and whole-genome sequencing (WGS) routinely generate tens of thousands of candidate variants per patient, rendering manual curation clinically impractical [1]. Phenotype-driven variant prioritization has emerged as an indispensable computational strategy to focus clinical attention on the subset of variants most consistent with the patient’s clinical presentation [2].
The foundation of this approach is the Human Phenotype Ontology (HPO), a rigorously curated, hierarchically structured vocabulary of phenotypic abnormalities that provides a standardized language for encoding clinical observations [1]. HPO terms underpin leading prioritization frameworks such as Exomiser and Genomiser, which apply ontology-aware similarity metrics to rank variants by their concordance with the patient’s phenotype profile [6].
However, the efficacy of phenotype-driven prioritization is directly contingent on the fidelity with which a patient’s clinical presentation is encoded in HPO. In routine clinical practice, phenotypic findings are predominantly recorded as unstructured free-text narratives, reflecting the diversity of clinical training, specialty-specific vocabulary, regional linguistic variation, and institutional documentation practices [3,4]. Bridging this gap, reliably translating heterogeneous clinical language into precise, machine-readable HPO identifiers continue to remain one of the most consequential bottlenecks in clinical genomics informatics.
This whitepaper describes the design, methodology, and clinical integration of the HPO Phenotype Matcher embedded within the Vgen23 clinical genomics platform. The system employs a hybrid AI pipeline that combines large language model (LLM)-driven clinical term extraction with vector-semantic similarity search against the HPO knowledge base. A structured clinician validation layer ensures phenotypic accuracy prior to seamless integration with variant annotation, ACMG/AMP pathogenicity classification, and gene-disease association frameworks, culminating in an explainable, phenotype-contextualized variant prioritization output.
2. Clinical and Scientific Background
2.1 The Human Phenotype Ontology
The Human Phenotype Ontology is a formally structured, controlled vocabulary developed and maintained by the HPO Consortium for the systematic description of phenotypic abnormalities in human disease [1]. Each concept is assigned a unique identifier (e.g., HP:0001250 for Seizure) and is embedded within a directed acyclic graph (DAG) that encodes hierarchical is-a relationships, allowing both specific and generalized phenotypic representations.
HPO enables cross-dataset interoperability through alignment with major biomedical resources including OMIM, Orphanet, ClinVar, and DECIPHER. As of 2025, HPO encompasses more than 17,000 terms spanning neurological, metabolic, skeletal, ophthalmological, and systemic phenotype domains, with continuous curation updates driven by literature synthesis and community contribution [1].
The ontological structure of HPO provides computational advantages that simple keyword vocabularies cannot replicate: semantic similarity metrics can quantify phenotypic relatedness using information-content-weighted graph traversal (e.g., Resnik, Lin, or Jiang-Conrath measures), enabling robust phenotype-to-disease matching even when the patient’s documented phenotype is less specific than the canonical disease description [2,6].
2.2 Phenotype-Driven Variant Prioritization
Phenotype-driven prioritization integrates a patient’s HPO profile with curated gene-disease-phenotype associations derived from databases such as OMIM, Orphanet, and ClinGen. Candidate variants are scored by computing the semantic similarity between the patient’s phenotype set and the HPO annotations of the gene(s) implicated by each variant. This score is then combined with pathogenicity evidence that is typically structured according to ACMG/AMP variant classification guidelines and inheritance pattern consistency to generate a composite prioritization rank [2,6].
Empirical benchmarking has consistently demonstrated that the accuracy of phenotype-driven prioritization is exquisitely sensitive to the quality of HPO input. Cooperstein et al. (2025) demonstrated that the inclusion of accurate, specific HPO terms significantly improves diagnostic ranking, whereas incomplete or non-specific annotations markedly reduce performance [6]. This dependency establishes HPO mapping quality as the primary determinant of diagnostic yield in phenotype-driven clinical genomic workflows.
2.3 The HPO Mapping Challenge
Despite the centrality of HPO in clinical genomics, reliable automated mapping of clinical narratives to HPO remains technically challenging. Several interrelated factors contribute to this difficulty:
- Terminological heterogeneity: Identical phenotypes are described using discipline-specific jargon, lay terminology, abbreviations, and synonym variants (e.g., “fits,” “convulsions,” “epileptic episodes” all mapping to HP:0001250).
- Contextual ambiguity: Negated or uncertain findings (“no seizures,” “possible intellectual disability”) require nuanced interpretation to avoid erroneous HPO assignment [3,4].
- Multilingual clinical environments: Clinicians in non-English-speaking regions may intermix native-language descriptors within otherwise English clinical notes, challenging monolingual NLP pipelines.
- Ontological granularity mismatches: Clinical documentation often captures phenotypes at a level of specificity that does not directly correspond to a single HPO leaf node, necessitating intelligent traversal of the ontological hierarchy [4].
- Incomplete ICD-to-HPO mappings: Existing crosswalks between ICD-10-CM and HPO via UMLS exhibit limited coverage for rare disease phenotypes, particularly for recently described conditions [5].
Existing NLP-based approaches including dictionary-based taggers, machine learning classifiers, and general-purpose large language models have demonstrated partial success in symptom extraction but inconsistent performance in downstream HPO identifier resolution [4,5]. Several machine learning approaches for automated Human Phenotype Ontology term recognition from clinical narratives have been previously explored [15]. These limitations motivate the development of purpose-built, ontology-grounded phenotype mapping systems for clinical genomic applications.
3. The Vgen23 HPO Phenotype Matcher: Architecture and Methodology
The HPO Phenotype Matcher is a multi-stage AI pipeline integrated natively within the Vgen23 clinical genomics platform. The system is designed to process realistic, noisy clinical inputs and produce high-precision HPO mappings that are validated before downstream use. The pipeline comprises five computational stages followed by a clinician validation interface.
| Step | Module | Description | Output |
|---|---|---|---|
| 1 | Clinical Term Extraction | LLM-driven NLP processing of free-text clinical notes; normalization of heterogeneous terminology | Candidate phenotype terms |
| 2 | Vectorization | Embedding of extracted terms into a shared semantic vector space aligned with the HPO corpus | Term embeddings |
| 3 | Similarity-Based Retrieval | Approximate nearest-neighbour search against pre-computed HPO embeddings using cosine similarity | Top-N HPO candidates per term |
| 4 | Disambiguation & Ranking | Similarity thresholding and context-aware re-ranking to resolve one-to-many term ambiguity | Ranked HPO ID list |
| 5 | User Validation | Interactive UI layer for clinician review and correction of mapped HPO terms | Validated HPO profile |
| 6 | Variant Prioritization | Ontology-aware phenotype similarity scoring integrated with ACMG/AMP pathogenicity evidence | Ranked variant shortlist |
Table 1. HPO Phenotype Matcher pipeline overview.
3.1 Stage 1: Clinical Term Extraction
The pipeline accepts free-text clinical inputs from the Vgen23 case submission interface. Input is processed by a fine-tuned large language model conditioned on a structured biomedical prompt that instructs extraction of all phenotype-relevant clinical concepts, their normalization to canonical English terminology, and the flagging of negated or uncertain findings. The model is specifically prompted to disambiguate synonyms, expand abbreviations, and handle multilingual embedding within clinical text.
“The patient has high blood sugar levels. There is a significant delay in overall development. The patient shows features suggestive of autism, including difficulties with social interaction and communication, along with repetitive behaviors or restricted interests.”
From this input, the extraction stage identifies candidate phenotypic terms such as hyperglycemia, global developmental delay, and autism spectrum disorder, appropriately normalized for downstream embedding.
3.2 Stage 2: Semantic Vectorization
Extracted clinical terms are encoded as dense vector representations using a biomedical sentence encoder trained on HPO definitions, synonyms, and phenotype-disease narratives. Critically, the same encoder is applied to pre-compute embeddings for all HPO terms, including official labels, hierarchical synonyms, and textual definitions, these are indexed in a high-dimensional vector database optimized for approximate nearest-neighbour (ANN) retrieval.
This shared embedding space enables semantic similarity computation beyond lexical overlap, allowing the system to retrieve ontologically appropriate HPO terms even when the clinical terminology deviates substantially from HPO canonical nomenclature.
3.3 Stage 3: Similarity-Based HPO Retrieval
For each extracted clinical term, the system executes an ANN query against the HPO vector index, returning the top-N candidate HPO terms ranked by cosine similarity. Similarity scores are calibrated against empirical thresholds derived from HPO-annotated clinical corpora to distinguish high-confidence mappings from low-confidence candidates requiring further disambiguation.
3.4 Stage 4: Disambiguation and Ranking
A single clinical term may return multiple plausible HPO candidates, a consequence of ontological synonymy, term polysemy, or hierarchical ambiguity. The disambiguation module applies a combination of similarity score thresholding, ontological hierarchy constraints (preferring more specific leaf nodes over broad parent terms when evidence supports specificity), and contextual coherence scoring across the full set of extracted terms to produce a ranked, deduplicated HPO mapping.
3.5 Stage 5: User Validation Layer
Recognizing that automated systems operating on heterogeneous clinical data cannot achieve perfect accuracy, the Vgen23 platform incorporates a structured clinician-in-the-loop validation step. The ranked HPO mappings including HPO identifier, preferred label, synonyms, and confidence score are presented in the case submission interface. Clinicians may accept, reject, modify, or supplement individual mappings before finalizing the phenotype profile. The workflow is designed to incorporate a clinician validation step prior to downstream analysis, ensuring that computational outputs are grounded in clinical judgment.
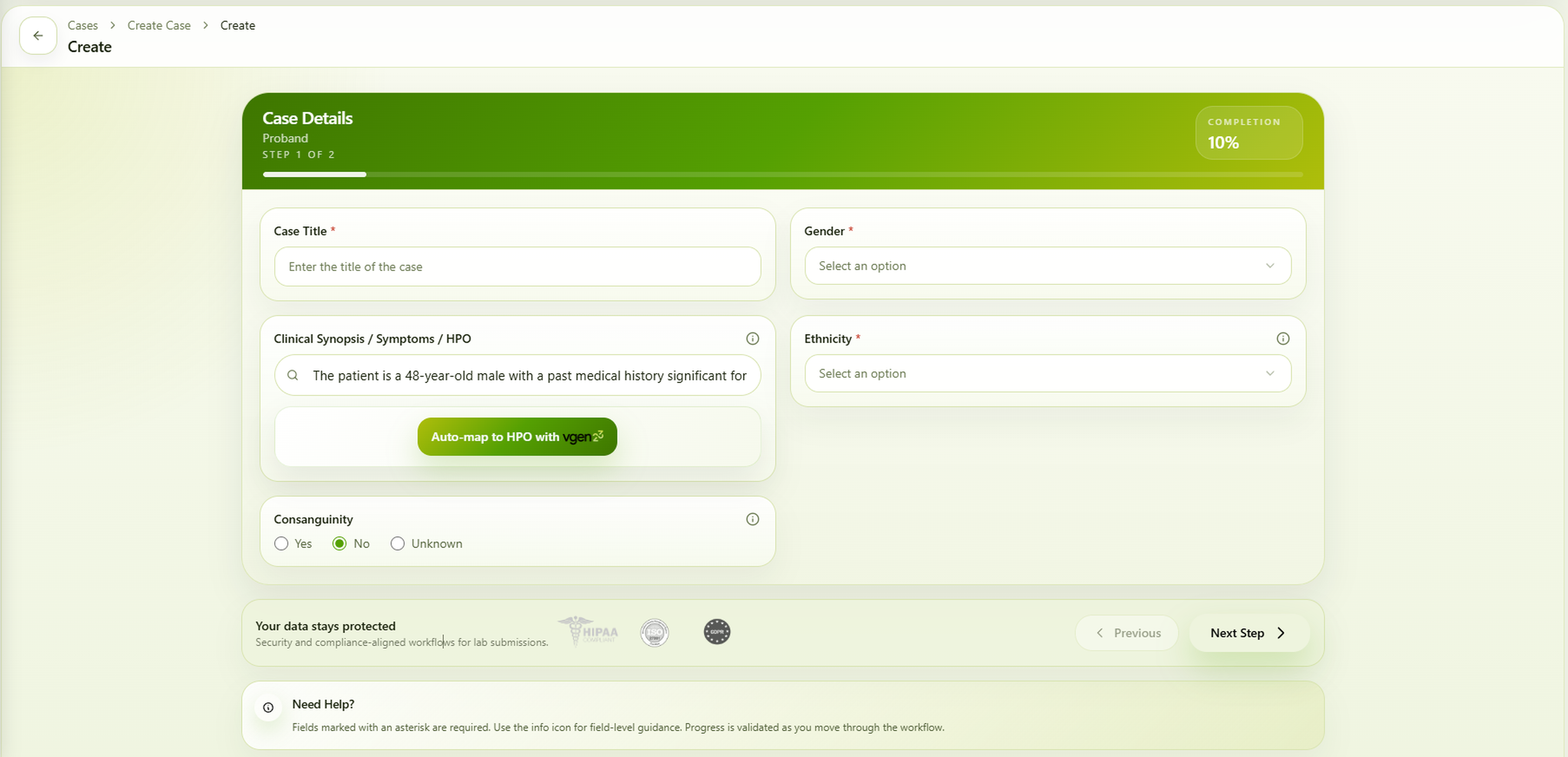
Clinical Input:
“The patient has high blood sugar levels. There is a significant delay in overall development, with delayed achievement of developmental milestones including sitting, walking, speaking, and learning. The developmental delay is severe and affects multiple domains, including motor, communication, and adaptive functioning. Additionally, the patient demonstrates features suggestive of autism spectrum disorder, including impaired social interaction, communication difficulties, and repetitive or restricted behaviors.”
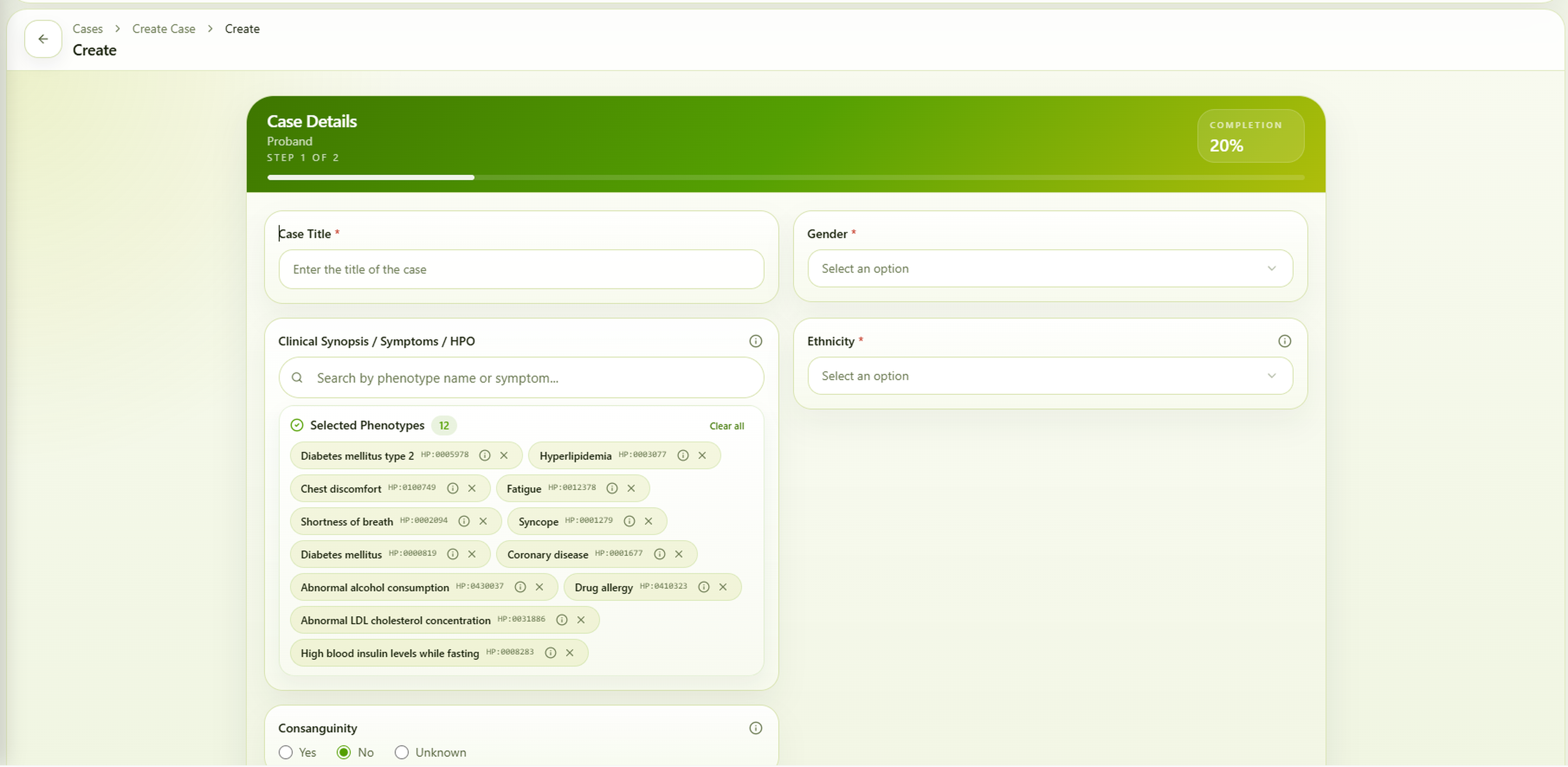
4. Performance Evaluation of the Vgen23 HPO Phenotype Matcher
To evaluate the effectiveness of the HPO Phenotype Matcher, we developed an AI-driven framework within the Vgen23 platform designed to improve rare disease phenotyping from unstructured clinical notes. The Vgen23 HPO Phenotype Matcher integrates Gemini-based large language models with a Retrieval-Augmented Generation (RAG) pipeline to identify and extract standardized Human Phenotype Ontology terms from clinical narratives.
The framework incorporates multiple prompting strategies, semantic retrieval, and ensemble decision-making to address the complexity and variability of rare disease phenotypes. Clinical phenotype descriptions generated by the language model are matched against HPO concepts using FAISS-based vector similarity search followed by cross-encoder semantic reranking.
The system was evaluated using the BioLarkGSC+ dataset [16], a high-quality expert-annotated benchmark corpus containing rare disease clinical narratives with gold-standard HPO annotations spanning multiple disease categories.
Among individual prompting strategies, the chain-of-thought approach achieved the best overall performance, with an F1-score of 0.560 and recall of 0.460, outperforming both zero-shot (F1 = 0.432) and few-shot prompting (F1 = 0.438). Ensemble-based majority voting improved prediction consistency, achieving an overall F1-score of 0.462. In contrast, weighted voting significantly increased precision to 0.862, although with reduced recall (0.256), highlighting a tradeoff between sensitivity and specificity.
| Pipeline | Precision | Recall | F1-Score |
|---|---|---|---|
| Zero-Shot Prompting | 0.820 | 0.290 | 0.432 |
| Few-Shot Prompting | 0.818 | 0.288 | 0.438 |
| Chain-of-Thought Prompting | 0.702 | 0.460 | 0.560 |
| Majority Voting Ensemble | 0.842 | 0.310 | 0.462 |
| Weighted Voting Ensemble | 0.862 | 0.256 | 0.394 |
Table 2. Performance comparison of prompting and ensemble strategies for HPO term extraction using the Vgen23 HPO Phenotype Matcher on the BioLarkGSC+ dataset
Overall, these results demonstrate that the Vgen23 HPO Phenotype Matcher can reliably extract clinically relevant HPO terms from unstructured clinical notes by combining prompt diversity, semantic reranking, and ensemble-based reasoning strategies.
5. HPO-Based Phenotype Profiling
Upon clinician validation, the curated HPO term set is transformed into a structured computational phenotype profile. The profile is constructed as a weighted vector over the HPO DAG, where individual term weights reflect confidence scores from the mapping pipeline, clinical relevance annotations provided by the clinician, and information-content values derived from HPO annotation statistics.
The system further augments the primary profile by incorporating ancestor terms via ontological propagation, a standard approach in phenotype similarity computation that improves recall by ensuring that similarity scoring algorithms can match at multiple levels of ontological specificity [2]. For example, a patient annotated with HP:0001250 (Seizure) automatically inherits annotations for HP:0012638 (Abnormal nervous system physiology) and related parent nodes, broadening the effective phenotype surface for gene-disease matching.
This structured phenotype encoding provides a consistent, machine-readable representation of the patient’s clinical presentation that can be reproducibly compared across patients, cohorts, and gene-disease knowledge bases, forming the computational substrate for phenotype-driven variant prioritization.
6. Challenges, Limitations, and Mitigations
6.1 Known Limitations
Despite the advances described in this whitepaper, several challenges inherent to clinical phenotype processing remain:
- Mapping Ambiguity: Vague, overlapping, or context-dependent clinical descriptors may produce low-confidence or multiple plausible HPO mappings, requiring clinician review and validation. Phenotype Incompleteness: Underreported, missing, or partially documented clinical features that are common in routine clinical practice can reduce the sensitivity and accuracy of phenotype-driven prioritization.
- Knowledge Base Coverage: Existing gene-phenotype association databases are biased toward well-characterized disorders, while ultra-rare, recently described, or poorly studied conditions may have limited phenotype annotations.
- Temporal Phenotype Evolution: Patient phenotypes may change over time due to disease progression, age-dependent manifestations, or treatment effects, necessitating periodic phenotype updates and reanalysis.
- LLM Hallucination Risk: LLM-based extraction pipelines may occasionally infer or generate clinically irrelevant phenotype terms from ambiguous text inputs. This risk is mitigated through ontology-based filtering and clinician validation workflows.
- Evaluation Scope: The current evaluation primarily focuses on phenotype extraction and HPO normalization performance. Prospective validation of end-to-end clinical diagnostic yield across diverse patient cohorts remains an area for future investigation.
6.2 Mitigation Strategies in Vgen23
The Vgen23 platform addresses these limitations through several complementary mechanisms:
- Vector Semantic Matching: Embedding-space retrieval extends beyond keyword matching, capturing synonymous and semantically proximate terms and improving mapping recall.
- Confidence-Scored Outputs: Similarity scores are surfaced to clinicians at the validation stage, enabling prioritized review of uncertain mappings.
- Hierarchical Phenotype Propagation: Ontological ancestor augmentation compensates for phenotype specificity mismatches and knowledge base annotation gaps.
- Continuous Reanalysis: The platform supports triggered or periodic reanalysis as phenotype profiles are updated or new gene-disease associations are published.
- Mandatory Validation Gate: The clinician validation step is architecturally enforced, ensuring no automated output reaches downstream analysis without human oversight.
7. Discussion
The clinical utility of phenotype-driven variant prioritization is fundamentally dependent on the quality of phenotype encoding. As clinical genomics advances toward population-scale sequencing and large-scale rare disease diagnostic programs, the demand for scalable and accurate phenotype normalization will continue to increase. In real-world clinical practice, however, heterogeneous inputs, multilingual documentation, and imprecise clinical narratives are common, creating substantial challenges for reliable computational phenotyping.
The Vgen23 HPO Phenotype Matcher is designed specifically to address these challenges through a hybrid AI architecture that combines large language model (LLM)-based clinical language understanding with vector-semantic retrieval against the Human Phenotype Ontology knowledge base. LLM-driven extraction enables flexible interpretation and normalization of heterogeneous clinical terminology, while ontology-grounded vector retrieval supports deterministic mapping to standardized HPO identifiers. The clinician validation layer further ensures that clinical expertise remains central to the final phenotype representation, preserving interpretability and clinical reliability within the diagnostic workflow.
Future directions include prospective clinical validation across diverse rare disease cohorts, integration of structured EHR-derived phenotype data, expansion of multilingual and longitudinal phenotype understanding, and incorporation of federated learning and real-world clinical feedback mechanisms to continuously improve phenotype mapping accuracy, robustness, and scalability.
8. Conclusion
The Vgen23 HPO Phenotype Matcher addresses a critical and long-standing bottleneck in clinical genomics: the reliable and scalable translation of heterogeneous clinical narratives into standardized Human Phenotype Ontology representations. By combining LLM-based clinical term extraction, vector-semantic similarity retrieval, ontology-aware disambiguation, and clinician-guided validation, the system provides a robust and explainable phenotype normalization framework.
When integrated with comprehensive variant annotation and phenotype-contextual ACMG/AMP interpretation within the Vgen23 platform, this framework enables clinically grounded phenotype-driven variant prioritization that is both transparent and actionable. By incorporating phenotype-aware interpretation into downstream genomic analysis, the platform supports more context-aware prioritization of clinically relevant variants while maintaining explainability throughout the workflow.
As clinical genomics advances toward large-scale precision medicine and population-wide sequencing initiatives, robust and explainable phenotype normalization will become increasingly central to genomic interpretation. The Vgen23 HPO Phenotype Matcher is designed to support this transition by enabling scalable, phenotype-aware, and clinically contextualized genomic interpretation workflows.
The Vgen23 platform is intended as a clinical decision-support and research-use system designed to assist, rather than replace, expert clinical interpretation.
References
- Köhler S, Gargano M, Matentzoglu N, et al. The Human Phenotype Ontology in 2021. Nucleic Acids Research. 2021;49(D1):D1207–D1217. doi:10.1093/nar/gkaa1043.
- Köhler S, Doelken SC, Mungall CJ, et al. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. American Journal of Human Genetics. 2008;83(5):610–615. doi:10.1016/j.ajhg.2008.09.017.
- Robinson PN, Köhler S, et al. Encoding clinical data with the Human Phenotype Ontology for computational differential diagnostics. Human Mutation. 2019;40(11):1855–1865. doi:10.1002/humu.23840.
- Zhang X, Chiu YC, Liang W, et al. Doc2HPO: a web application for efficient and accurate HPO concept curation. Nucleic Acids Research. 2019;47(W1):W566–W570. doi:10.1093/nar/gkz386.
- Searle T, et al. Implications of mappings between ICD clinical diagnosis codes and the Human Phenotype Ontology for phenotypic analyses. Journal of the American Medical Informatics Association. 2024. doi:10.1093/jamia/ocae133.
- Cooperstein IB, Marwaha S, Ward A, et al. An optimized variant prioritization process for rare disease diagnostics: recommendations for Exomiser and Genomiser. Genome Medicine. 2025;17(1):127. doi:10.1186/s13073-025-01546-1.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine. 2015;17(5):405–424. doi:10.1038/gim.2015.30.
- Robinson PN. Deep phenotyping for precision medicine. Human Mutation. 2012;33(5):777–780. doi:10.1002/humu.22080.
- Birgmeier J, Deisseroth CA, Hayward LE, et al. AMELIE speeds Mendelian diagnosis by matching patient phenotype and genotype to primary literature. Science Translational Medicine. 2020;12(544):eaau9113. doi:10.1126/scitranslmed.aau9113.
- Haendel MA, Chute CG, Robinson PN. Classification, Ontology, and Precision Medicine. New England Journal of Medicine. 2018;379(15):1452–1462. doi:10.1056/NEJMra1615014.
- Pesquita C, et al. Semantic similarity in biomedical ontologies. PLoS Computational Biology. 2009;5(7):e1000443.
- Johnson J, Douze M, Jégou H. Billion-scale similarity search with GPUs. IEEE Transactions on Big Data. 2019;7(3):535–547.
- Devlin J, Chang MW, Lee K, Toutanova K. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. NAACL-HLT. 2019.
- Liu H, et al. Deep learning based phenotype extraction from clinical narratives for rare disease diagnosis. Database (Oxford). 2019;2019:baz129. doi:10.1093/database/baz129
- Lobo, Manuel, Lamurias, Andre, Couto, Francisco M., Identifying Human Phenotype Terms by Combining Machine Learning and Validation Rules, BioMed Research International, 2017, 8565739, 8 pages, 2017. https://doi.org/10.1155/2017/8565739.
- Groza T, Köhler S, Moldenhauer D, et al. The Bio-Lark benchmark corpus for the recognition of Human Phenotype Ontology concepts in clinical text. Database (Oxford). 2015; 2015: bav005. doi:10.1093/database/bav005
List of Abbreviations
| ACMG | American College of Medical Genetics and Genomics |
| AMP | Association for Molecular Pathology |
| ANN | Approximate Nearest Neighbour |
| DAG | Directed Acyclic Graph |
| EHR | Electronic Health Record |
| HPO | Human Phenotype Ontology |
| LLM | Large Language Model |
| NGS | Next-Generation Sequencing |
| NLP | Natural Language Processing |
| OMIM | Online Mendelian Inheritance in Man |
| VCF | Variant Call Format |
| WES | Whole-Exome Sequencing |
| WGS | Whole-Genome Sequencing |